Research Projects 🔬
Click on the titles to read more.
Information Theory of Composite Sequence Motifs 🧬
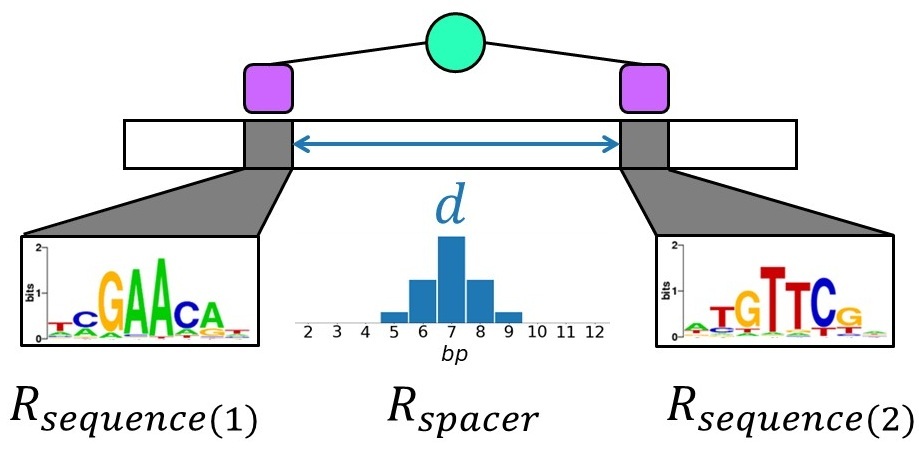
Through nucleotide conservation, the genomic positions that must be bound by transcription factors can provide information.
The information content (named Rsequence) can be measured (in bits) and happens to be nearly equal to the amount of information theoretically required for the target location to be accomplishable (a quantity named Rfrequency)
(Schneider et al., 1986).
I proposed a generalized framework for this fundational theory that can be used to describe the evolution of composite sequence motifs of n elements
(Mascolo & Erill, 2024).
The classical theory by Schenider can be seen as a special case where n=1.
I also show how this framework can be applied to study different aspects of target recognition mediated by molecular complexes (how protein flexibility can co-evolve with spacer variability, the thermodynamic efficiency of different recruitment strategies, and the effect of mutation spectra on the evolvability of different information-encoding strategies).
📌 Open poster
FLEMINGO 🦩
I developed a motif discovery tool that can discover composite motifs (see Composite Sequence Motifs ) in biological data (sets of co-regulated promoters) through evolutionary computation. The tool, called FLEMINGO (for FLExible Motif INference via Genetic Optimization), can detect motifs encompassing spacers (or gaps) of variable length, as well as DNA shape features (inferred from the DNA sequence).
![]()
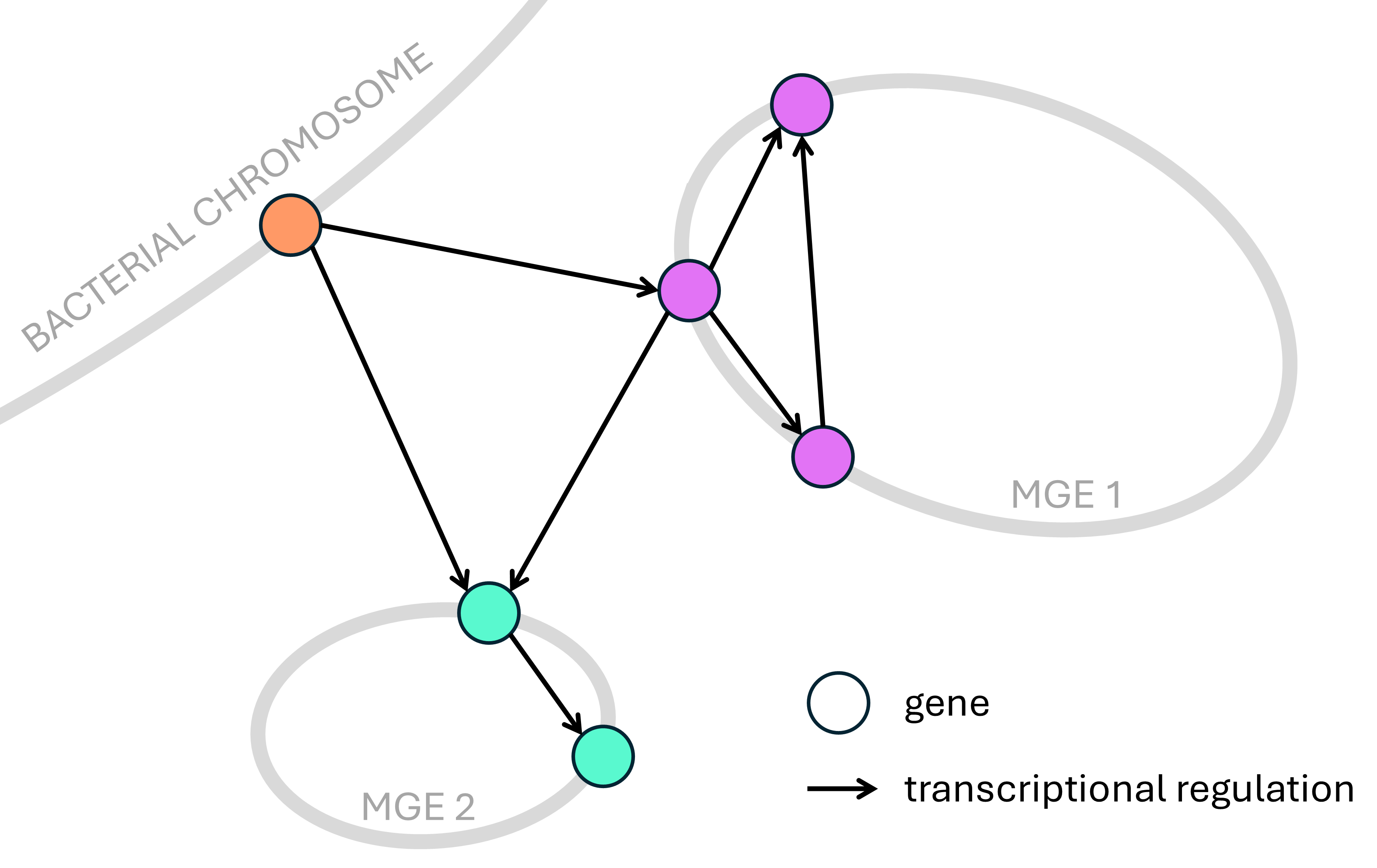
Cross-replicon gene regulation ⚙
In a transcriptional regulatory network (TRN), every node represents a gene, and every link represents a transcriptional regulation,
connecting a transcription factor with one of its targets.
Could there be hybrid TRNs comprising genes that are encoded on different replicons? For example, in bacterial cells we could find the bacterial genome, but also natural plasmids, as well as bacteriophage genomes and other mobile genetic elements (MGEs).
In 2022, we showed that many phylogenetically unrelated bacteriophages that infect Alphaproteobacteria have convergently evolved DNA binding sites for the host cell cycle regulator CtrA, suggesting that these bacteriophages synchronize lysis with the life stage of the host to maximize their infective yield, through a pathway that we termed lytic deferment.
(Mascolo et al., 2022).
I developed an algorithm to systematically predict the presence of such hybrid TRNs. This method can re-discover the few cases that are already known in the literature, but it also finds many new realistic candidates, revealing that hybrid TRNs are highly prevalent in the microbial world (unpublished work). Several of its predictions are currently being experimentally validated.
📰 Media coverage on host-phage cross-regulation
📌 Open poster
Viral binding 👾
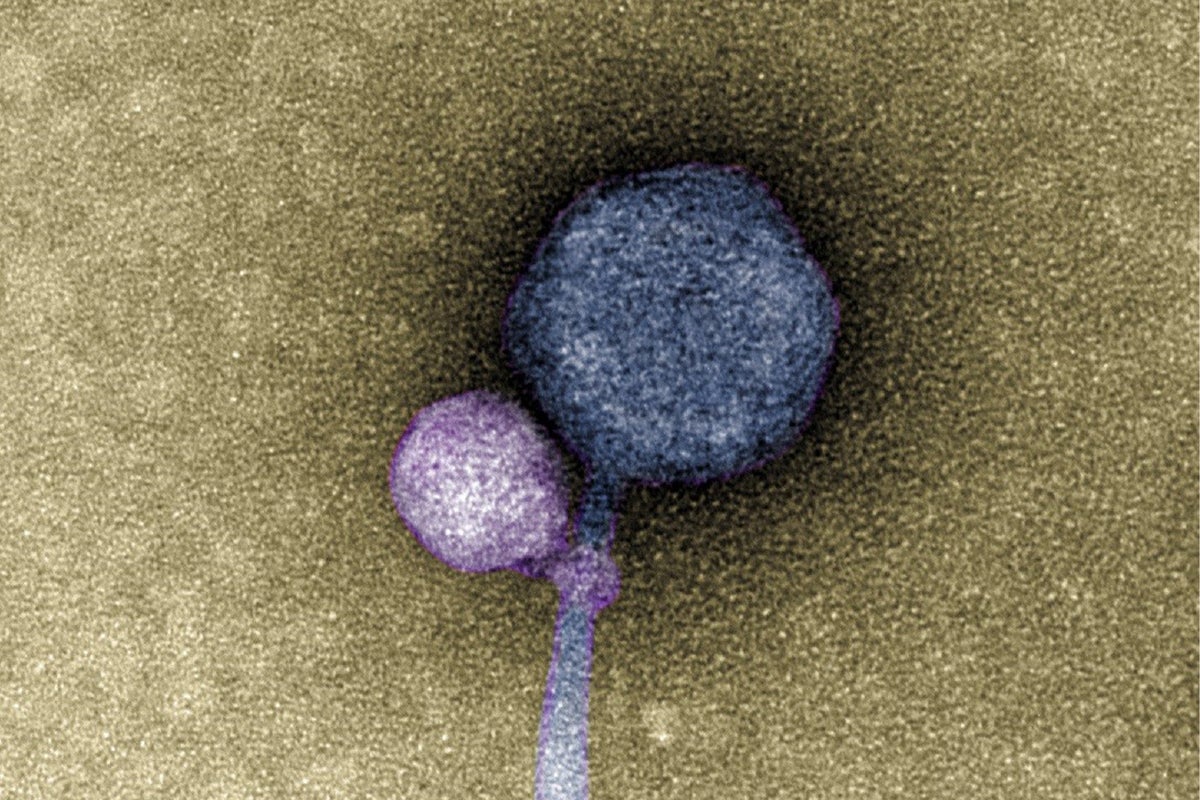
In a work where I share co-first authorship with Tagide deCarvalho, we describe for the first time a virus that attaches itself to another virus. Tagide deCarvalho discovered this novel type of interaction while observing under the electron microscope (TEM) a novel bacteriophage that our students named MindFlayer. She noticed that most of them had a smaller virus attached to the "neck" (the part that connects the head, or capsid, to the tail). This explained what earlier, during the genome sequencing of MindFlayer, seemed to be a systematic contamination. With the microscope, she revealed that the shorter secondary sequence was not a contamination but the genome of this smaller hitchhiker, which we named MiniFlayer. I performed a bioinformatics analysis, showing that MindFlayer and MiniFlayer have been co-evolving for a long time, and further suggesting that MiniFlayer exploits the genes of MindFlayer. However, unlike previously known interactions, it doesn't wait in the host cell for MindFlayer to co-infect. Rather, it ensures the presence of the genes it needs by entering the host together with MindFlayer.
📰 Media coverage on MindFlayer and MiniFlayer
Publications
Here you can find an up-to-date list of my publications.